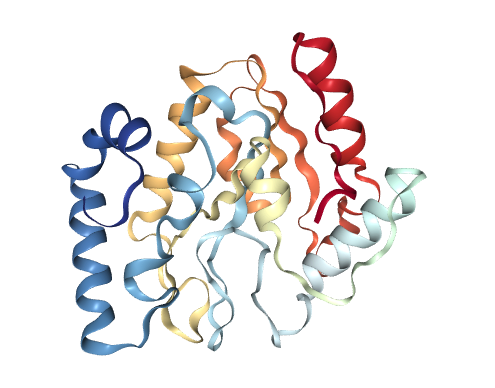
Through my interest for Cystic Fibrosis (if you haven’t seen them yet then check out The Frey Life for more information on what life with Cystic Fibrosis can look like) I have focused on understanding what the problem with misfolded proteins is all about and what the difficulties are in inventing new medicine to improve the life of patients living with an illness that is caused by proteins not working correctly. The problem in Cystic Fibrosis is that the proteins which act as channels for chlorid ions aren’t constructed correctly and hence cannot perform their job, either because they are destructed before they are build into the cell membrane or because they don’t function (well) when they get there. What surprised me when starting to learn about the mechanisms is the fact that only a single error in a sequence of thousands of amino acids can cause the illness. To illustrate this, here is the 3D-model of the CFTR-protein with the 508th amino acid marked. That amino acid is missing in most Cystic Fibrosis patients (but not all, there are more than thousand mutations) and that causes the protein to be destructed by quality mechanisms in the cells. Small change, big effect.

I like being naive about unsolved challenges. I know they are typically unsolved for a reason, but not trying anyway will mean they will not be solved. One of those challenges is predicting the 3D-model of a protein from the amino acid sequence. The hope is, that the amino acid sequence alone is information enough to predict the 3D-structure. I have a feeling that the surface of ribosomes are important as well, but only time will tell if that plays a role. In 2017 I was to a conference on using machine learning for improving simulation of molecule structures (an protein structures as the most important special case of that) called Machine Learning and Molecules. The main takeaway for me was that we haven’t got very far yet. Yes, the simulation time could be improved, but we are still not able to simulate interesting proteins for a time range that would be helpful. Currently weeks and months are still needed for even small molecules.
Especially because we haven’t got far in prediction of protein structure from the amino acid sequence I have chosen to make this my new long-term project. I currently experiment with the reverse problem. Taking in the 3D-structure and predicting the amino acid sequence. My input is a distance map, where I take the 3D-structure from the PDB database, a universally recognized and used database containing the 3D-structures of molecules found experimentally using X-ray diffraction and Nuclear Magnetic Resonance techniques. The distance map represents the distance between the alpha-carbon atoms in the amino acids. The close these are together, the darker the color of the pixel in the image. An example is shown below (https://www.rcsb.org/structure/1AKZ):

The reason why I chose a distance map is the translational and rotations invariance. I’m worried that if I would train a model to predict the 3D-coordinates of the alpha-carbon atoms, the learning would fail because the model predicts the atoms to be in a different location than the training data, but the location might be just right, if the molecule was just rotated differently. That problem is not present with the distance map. The distance map has just another problem, as one of my co-workers pointed out: I have an output space that scales with O(N^2), where N is the length of the input. That’s a lot of outputs!
My current idea is to build a few convolutional layers which combine a few neighboring amino acids and maps them into a lower dimension. Let me put it another way: Let’s say we combine 5 amino acids. The number of possible combinations is 20^5=3.2 million (because there are 20 amino acids). The first few layers should map this from a 3.2 million dimensional space into a space of a few thousand dimensions. Each amino acid should in this way, taking into account it’s neighbors, being mapped to a new node in an intermediate layer. The data from this layer can then pairwise be the input to a new network that can predict the distance map value. When the distance map is correctly predicted, the 3D-structure can be reconstructed using triangulation.
I’m still working on building the networks and learning to use TensorFlow. When I have any results ready I will post them as a new post.
Comments are closed.